The melanogenesis hypothesis
The whole thing started with an analogy. The final step of firefly luciferin biosynthesis, where Cys-HQ cyclizes into the benzothiazole ring, looks structurally similar to a reaction in melanin synthesis. In melanogenesis, laccases (a family of copper-dependent oxidases) catalyze the oxidative cyclization of similar phenolic substrates. Tyrosinase and laccases grab small molecules, rip electrons off them using copper chemistry, and force ring closures.
My hypothesis was simple: the missing luciferin synthase might be a laccase or laccase-like copper oxidase that was repurposed for luciferin production.
Step 1: Are laccases expanded in fireflies?
If laccases evolved a new role in luciferin biosynthesis, you might expect to see more laccases in fireflies than in non-luminous beetles. I ran BLAST searches comparing laccase gene families between Photinus pyralis (common eastern firefly) and Tribolium castaneum (red flour beetle).
The result: a roughly 3× expansion of laccase-family genes in the firefly. More laccases than a normal beetle needs.
Step 2: Mining Fallon’s RNA-seq data
Fallon’s lab had published a comprehensive RNA-seq dataset comparing gene expression in the firefly lantern (the light organ) vs. fat body tissue, covering all 15,773 predicted genes in the P. pyralis genome. I couldn’t search for “laccase” directly in the Gene Ontology annotations, so instead I filtered for the biochemical signature of a laccase: GO:0005507 (copper ion binding) combined with GO:0016491 (oxidoreductase activity).
That returned seven copper-binding oxidoreductases.
Step 3: One gene screamed from the data
Six of those seven were barely expressed in the lantern. TPM values between 0.5 and 4.1, doing normal beetle things like cuticle hardening. But PPYR_12315 had:
- A lantern TPM of 420.8
- 100× to 1,000× higher than the other copper oxidases
- 14.5× upregulated over fat body
- Ranked as the 85th most significantly differentially expressed gene out of over 15,000
I then BLASTed PPYR_12315 against every firefly species with genomic data on NCBI. Orthologs appeared across the entire Lampyridae family. The gene was conserved across lineages that diverged tens of millions of years ago, with identity levels (79–91%) indicating a functional protein under strong purifying selection. If every firefly species maintains this protein, it’s doing something important.
AlphaFold structure prediction
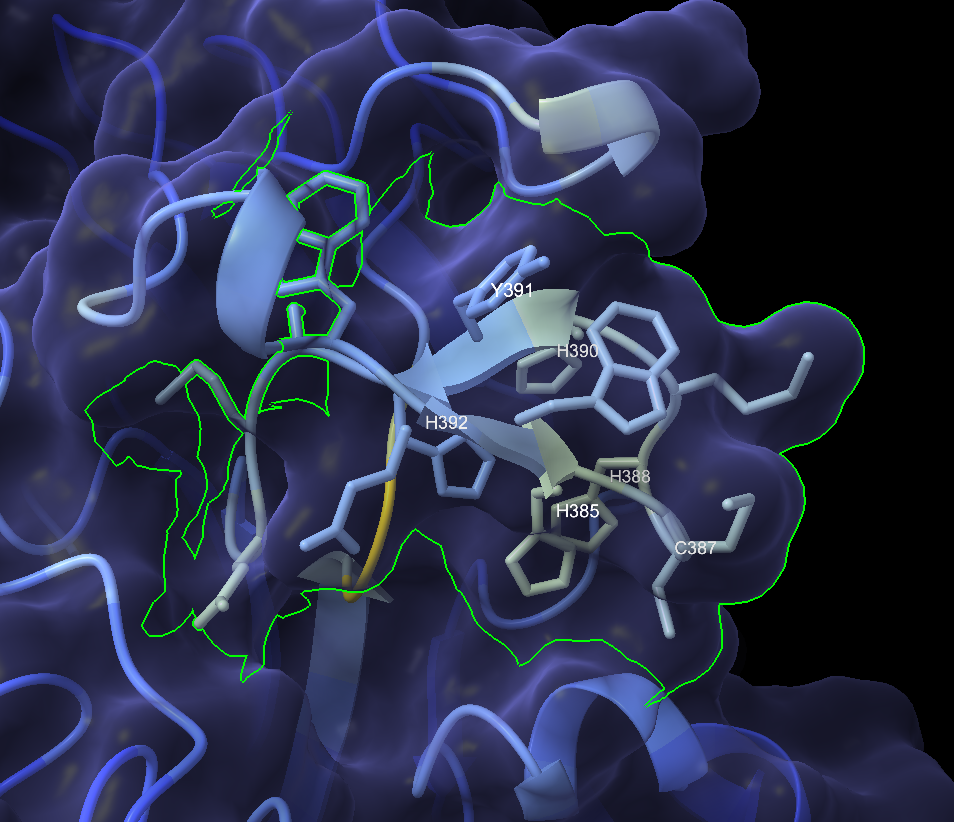
I ran the 488-amino-acid protein through ColabFold to get a predicted 3D structure, then loaded it into ChimeraX for analysis. The structure revealed dense clusters of histidine and cysteine residues: HIS 385, HIS 388, HIS 390, HIS 392, CYS 387, CYS 419arranged in a pattern consistent with a copper center.
The active site motif read W-E-W-H-M-C-H-M-H-Y-H across positions 382–392: four histidines packed around a putative copper coordination site.
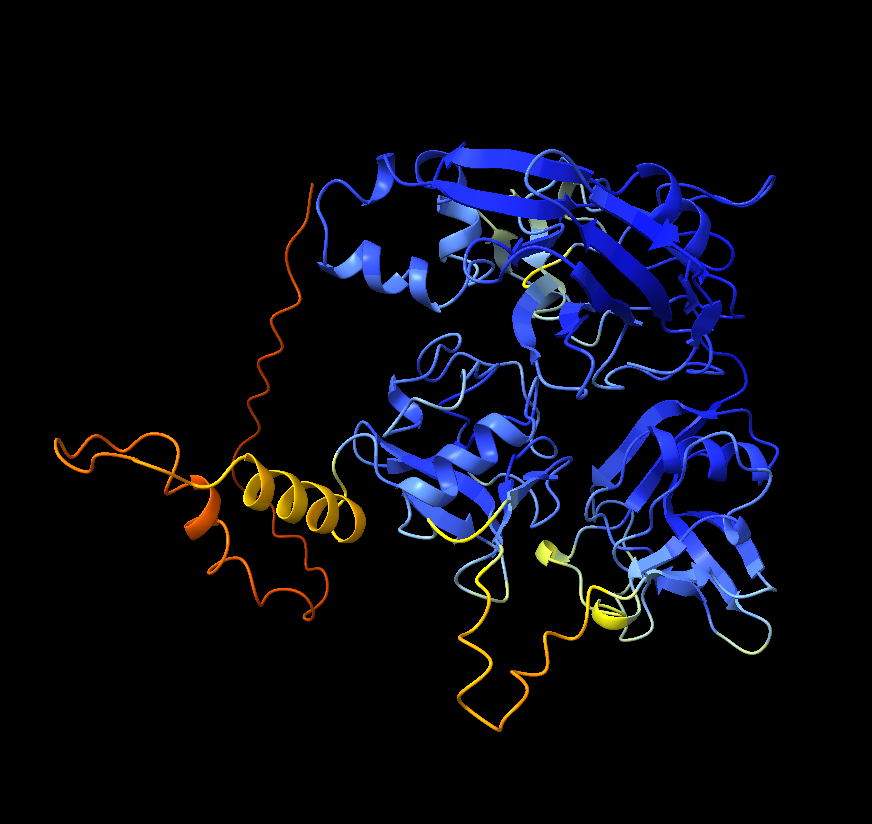
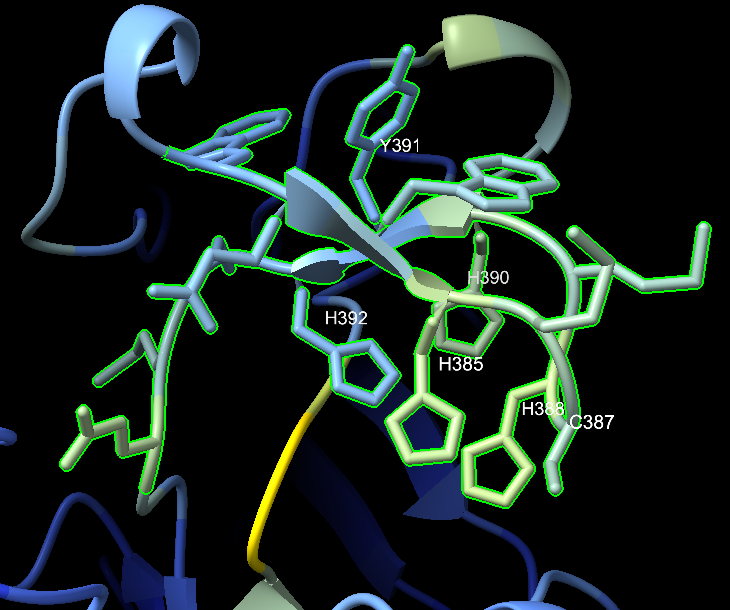
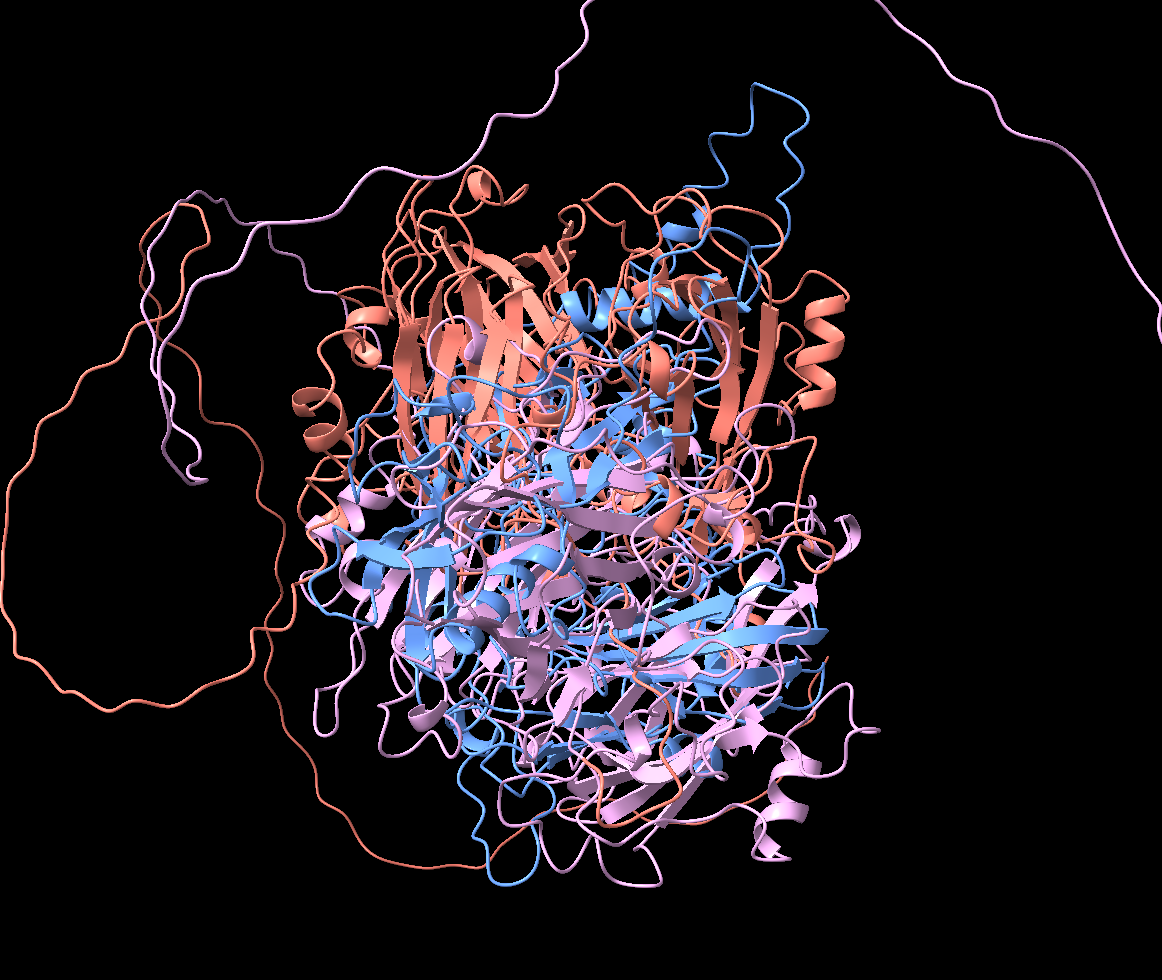
Structural comparison with a known laccase
I superimposed the predicted structure against TcLac2A, a characterized insect laccase from Tribolium castaneum. The overall RMSD was 31Å, meaning the global fold had diverged dramatically, but the copper-coordinating residue clusters were preserved.
My interpretation at the time: the catalytic engine is the same (copper oxidation chemistry), but the housing around it has been reshaped, potentially to accommodate a different substrate. I was already framing this as neofunctionalization.
Electrostatic surface mapping
I generated electrostatic surface potentials in ChimeraX and found a large negatively charged patch around the predicted active site. Since Cys-HQ carries a positive amine group on its cysteine moiety, a negatively charged surface could help attract and orient the substrate. Another piece of the puzzle that seemed to fit.
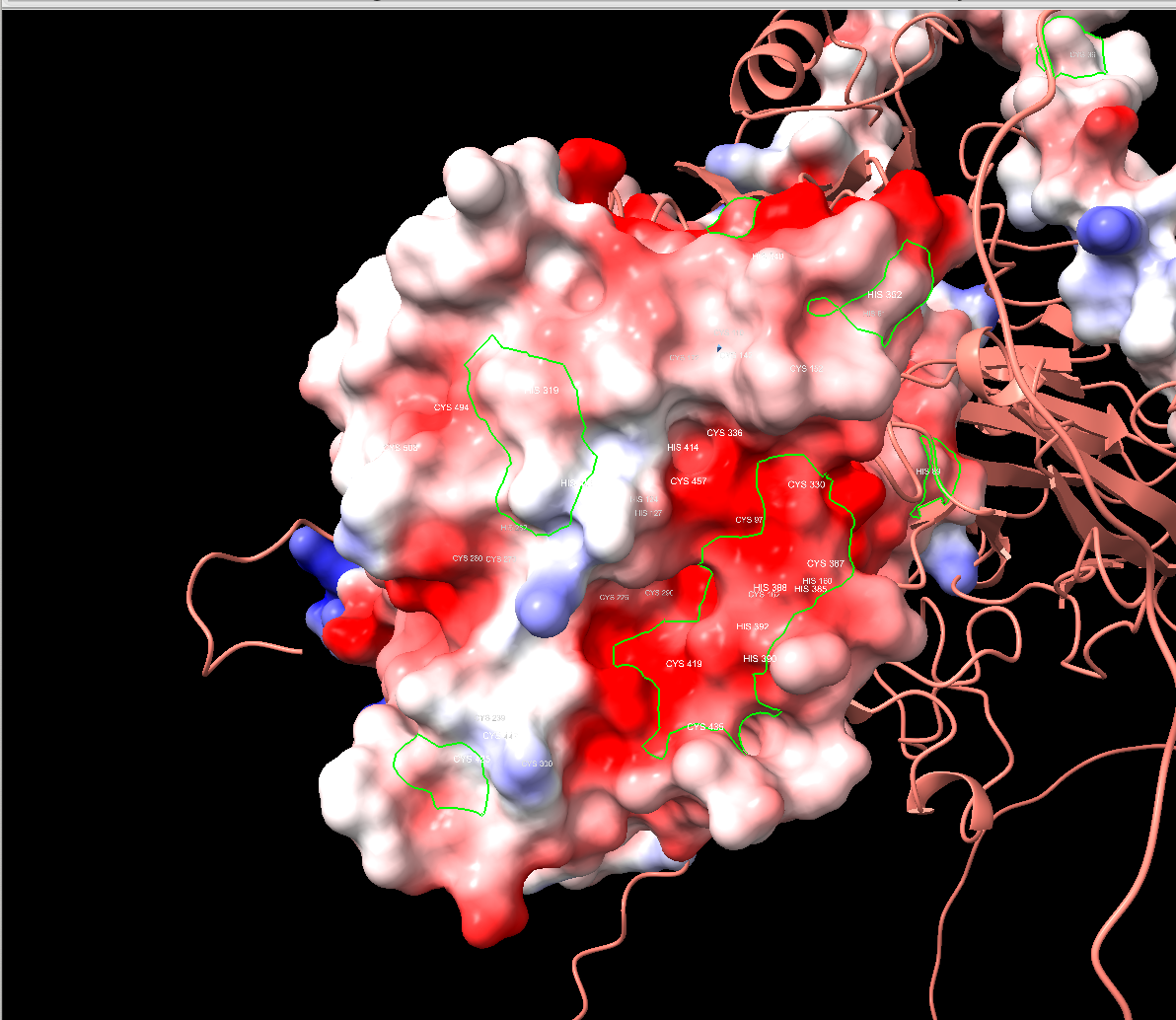
Geometric substrate fitting
I then checked whether benzoquinone and cysteine could physically fit in the active site cavity. They could, the pocket was large enough, and the copper center was positioned appropriately for substrate activation. At the time, I noted (correctly) that geometric fit is necessary but not sufficient. In hindsight, I should have weighed that caveat more heavily.
The first red flags
I pasted the N-terminal protein sequence into ChatGPT for a second opinion. Its analysis flagged several things I hadn’t fully reckoned with:
- A strong signal peptide at the N-terminus (
MKSEIITVVACLTVLVFPSFRA) — classic ER targeting, indicating the protein is secreted. This was not what you’d expect for a cytosolic small-molecule synthase. - Cysteine-rich repeat-domain architecture with patterns like
...GAICDDEW...and...CRFDGWGSHDCE..., typical of extracellular structural proteins, not compact cytosolic enzymes. - Basic clusters (
KHVKRLKKEE,VRVRLRGGRV) that looked like propeptide cleavage or processing regions—again, hallmarks of a secreted protein.
HMMER: the definitive answer
Scott suggested I run the protein through InterPro and HMMER hmmscan. HMMER doesn’t just match individual motifs the way my earlier InterPro analysis had; it compares the full statistical profile of the protein sequence against curated hidden Markov models for every known protein family.
The results were definitive:
| Pfam ID | Domain | E-value |
|---|---|---|
| PF01186 | Lysyl oxidase | 4.6e-97 |
| PF00530 | SRCR (scavenger receptor cysteine-rich) | 4.7e-30 |
| — | Cupredoxin / multicopper oxidase | No hits |
Zero cupredoxin or multicopper oxidase domain hits.
PPYR_12315 is a lysyl oxidase-like 2 (LOXL2) protein, not a laccase. The NCBI auto-annotation as LOXL2 had been correct the entire time. The E-value for the lysyl oxidase match was 4.6e-97, extremely confident. I ran it twice with both the 488 aa and 513 aa versions of the protein. Same result both times.
Final confirmation: unrestricted BLAST
I ran one more test: BLASTP against all of NCBI’s non-redundant protein database, organism unrestricted, to see the full phylogenetic picture. The results confirmed my suspicions. Lesson learned. In my other posts, you now see me BLAST and HMMER before anything else for a reason.
Firefly copies clustered together at 75–83% identity. But then non-luminous beetles appeared: Tenebrio molitor at 68%, Zophobas morio at 69%, Tribolium castaneum at 66%. The gene isn’t firefly-specific. Every beetle has one. It’s a normal insect LOXL2 ortholog that happens to be highly expressed in the lantern.
The gap between firefly and non-firefly identity (about 10–15 percentage points) is notable but well within normal evolutionary drift for an orthologous gene. This was not the missing enzyme.
What I learned
Motifs are not families
The biggest lesson: finding copper-binding motifs and cysteine-rich regions in a protein doesn’t tell you what the protein is. Multiple unrelated enzyme families use copper. InterPro and manual motif analysis can pick up superficial similarities. HMMER profile-based classification is the gold standard because it compares against the full statistical profile of each family, not just individual motifs. I should have run HMMER first, before everything else.
Beware of confirmation bias
I started with a hypothesis, found supporting evidence, and kept finding more. Expression data, cross-species conservation, AlphaFold structures, electrostatic maps, substrate fitting, everything pointed the same way because I was looking for reasons to believe. Each piece of evidence was real, but my interpretation was shaped by what I wanted to find. Although as I also learned, confirmation bias only goes so far in biology.
Though maybe I just wanted an excuse to use AlphaFold.
The project builds on Tim Fallon’s published genomic data for Photinus pyralis and represents original bioinformatics analysis performed by the author.